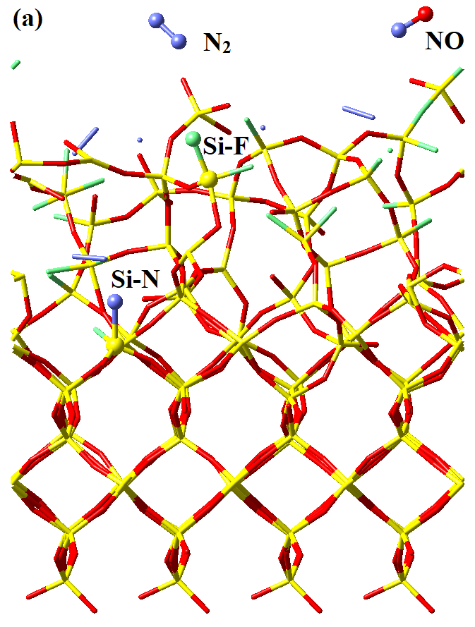
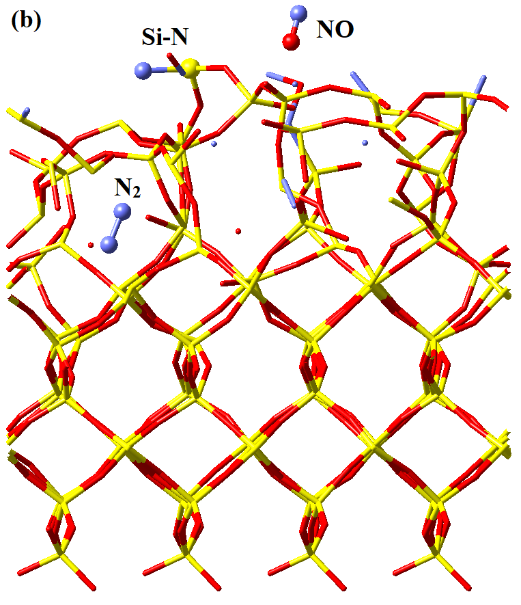
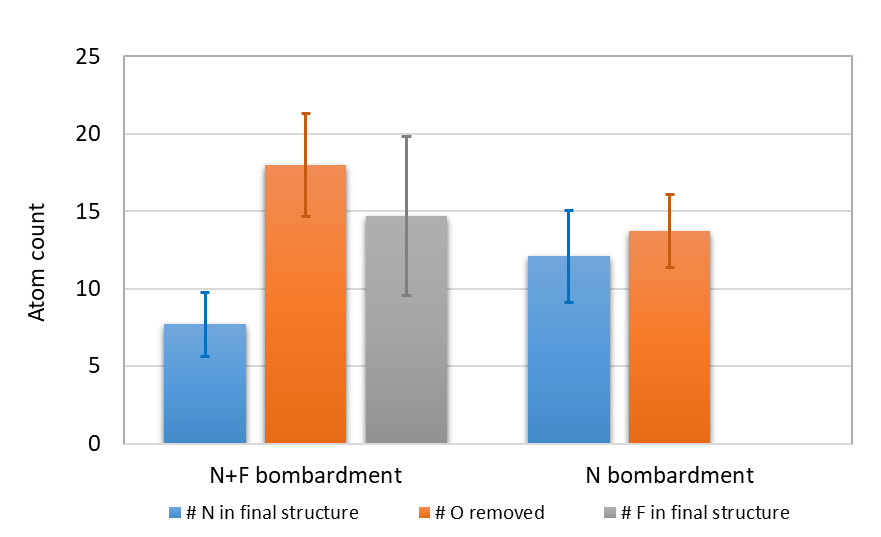
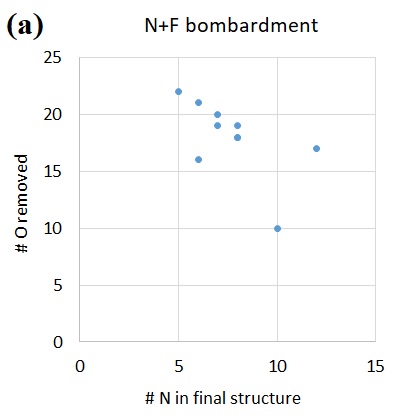
With a phenomenological understanding of surface reactions offered by the MD simulations, we now examine their underlying energetics using QC calculations in order to derive fundamental mechanistic insights. These static energetics calculations with QC not only yield the equilibrium structures of reaction intermediates and transition states, but also offer electronic structure information absent from MD simulations. Therefore, it is beneficial to cross-compare the MD and QC results for a better understanding of the etch mechanisms.
Figure 4.
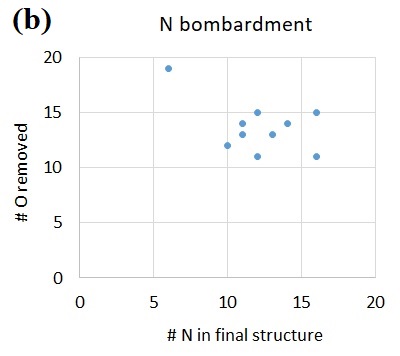
The reaction energetics for the surface dangling *Si-O attacked by N leading to NO abstraction. The most stable spin states are presented for each geometry. The potential energy sum for isoalted *Si-O (doublet) and N atom (quartet) is chosen as reference. Figure 5.
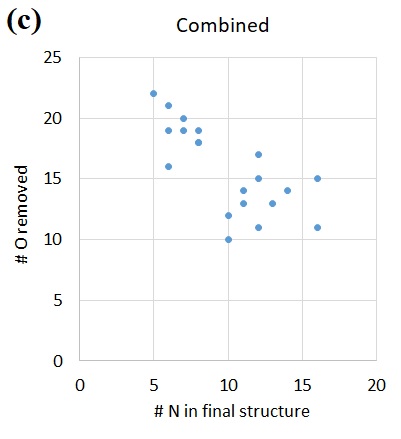
Geometries and potential energies of various possible configurations of surface adsorbed NO. The potential energy sum for isolated *Si-O (doublet) and N atom (quartet) is chosen as reference, consistently with Figure 4. The singlet *Si-O-N structure is unstable, which upon geometry optimization spontaneously re-organizes into singlet *Si-N-O. All 3 other structures are confirmed as local minima by the absence of imaginary vibrational frequencies. For an isolated doublet NO molecule in vacuum, the Mulliken spin densities on N and O are 0.719 e and 0.281 e, respectively. With the surface under continuous ion bombardment, the surface becomes amorphous, with many dangling bonds or under-coordinated atoms present. Examination of the surface structures in Figure 1, for instance, shows the abundance of -O dangling bonds (mostly *Si-O groups). The presence of these dangling bonds or under-coordinated atoms provides active sites for forming new chemical bonds with incoming species from the plasma, which eventually results in volatile product formation. Thus, the surface reactions and substrate volatilization in a plasma etch system are ion-enhanced, allowing for significantly lower process temperatures than plasma-less dry etch (which relies totally on the thermal fluctuations to overcome the activation energy barriers).
Given this, we may justifiably start with the model structure (Figure 4) for the surface *Si-O dangling bond (apparently the most abundant form of dangling bonds according to Figure 1). The most stable electronic state for the *Si-O group is a spin doublet, with 1 unpaired electron. After the attack by an incoming N (quintet) radical, a triplet *Si-O-N intermediate is formed, at a ΔE of -3.360 eV. Evidently, the N-O bond formation is energetically highly favorable. Subsequently, the triplet *Si-O-N undergoes a rotation of the NO adsorbate, breaking the Si-O bond and forming the Si-N bond instead, which slightly lowers the potential energy by another 0.263 eV. Eventually, the triplet *Si-N-O group releases the NO adsorbate in its doublet state, and an under-coordinated Si (bonded to 3 O atoms, also a doublet) is exposed. The NO elimination requires a potential energy increase by 1.857 eV, which can be readily afforded by the incoming ion bombardment in the nearby area and its associated local heating. Overall, the reactions from the initial N attack leading up to the desorption of NO has a favorable ΔE of -1.766 eV.
We now take another look at the adsorbed-NO intermediates, to further unravel the intricacies of the various possible configurations, both structural and electronic. Firstly, for both the *Si-O-N and the *Si-N-O structures, the triplet is lower in energy than the singlet (Figure 5). Second, for both the singlet and the triplet, the *Si-O-N structure is higher in energy than the *Si-N-O structure. Specifically, for singlet *Si-O-N, the structure is not a true local minimum, which upon geometry optimization spontaneously relaxes into the *Si-N-O singlet local minimum, at a ΔE of about -1.2 eV. This unstable singlet *Si-O-N structure may also undergo a spin flip to generate the triplet *Si-O-N structure, with about 1.3 eV decrease in potential energy. Finally, to comment on what structures may be involved during the N attack on O, we assume that the electronic and nuclear degrees of freedom are decoupled and that the incoming N and the surface *Si-O are both in their electronic ground state. In this case, the electronic state for a system of *Si-O and N atom approaching each other will opt for the lower-energy triplet as the groups gradually approach each other. This yields the *Si-O-N triplet structure as shown in Figure 4.
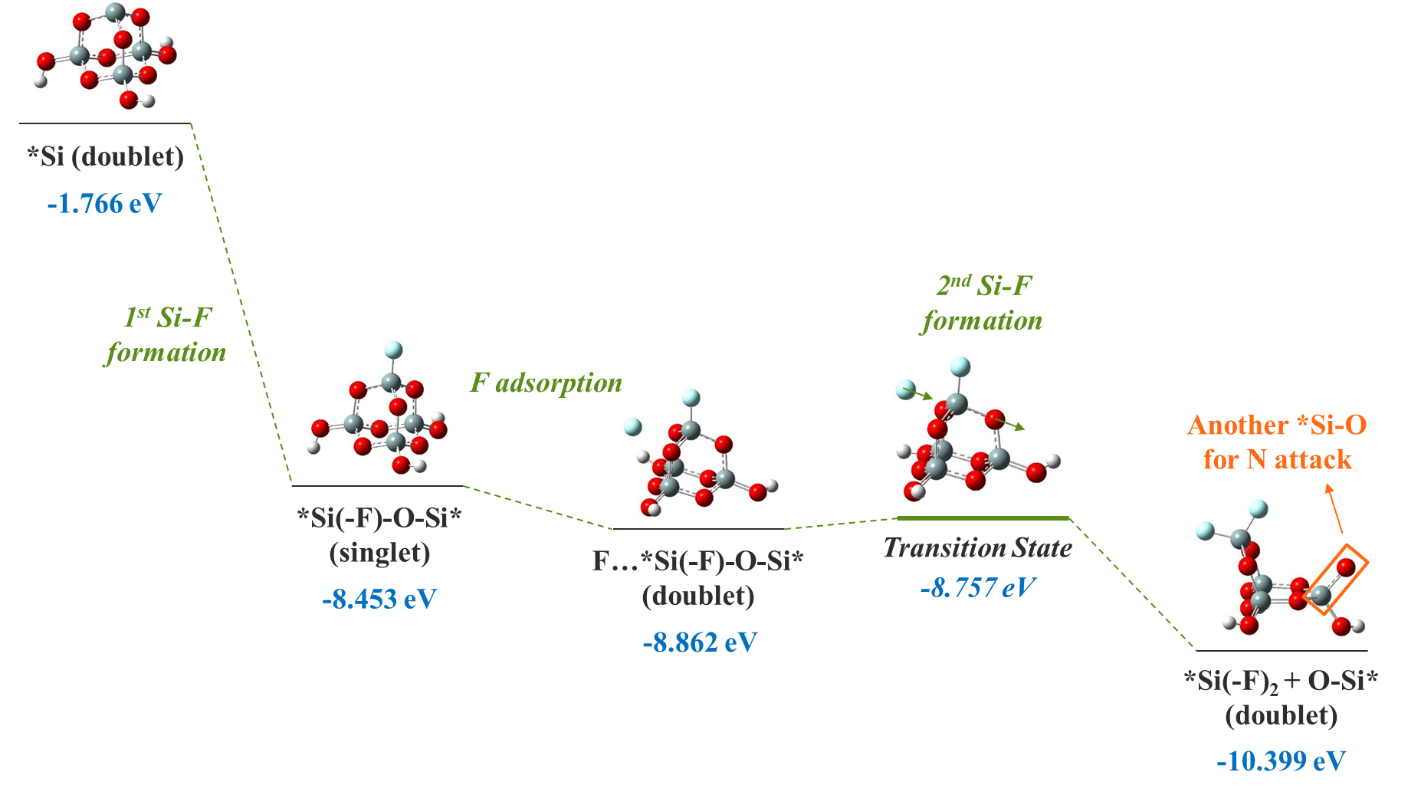
Following the NO desorption, the created 3-coordinated *Si group (doublet) becomes an excellent open site for F atom attack, forming Si-F bond (Figure 6) at a significant 6.687 eV potential energy drop. Then a second F atom approaches and becomes physisorbed, which again has a favorable ΔE of -0.409 eV. Subsequently, the Si-O-to-Si-F substitution reaction takes place via a transition state with a 0.105 eV activation energy, forming another Si-F bond and lowering the potential energy further by 1.537 eV. Further Si-O-to-Si-F substitutions ultimately leads to the formation of the SiF4 molecule as a major Si etch product. We also notice that each Si-O-to-Si-F substitution creates an additional *Si-O, which in essence is the reactant for N attack in Figure 4, thus completing a full circle of reactions driving the plasma-assisted SiO2 etching.
We summarize the QC findings in comparison with the MD results. Both QC and MD predict the N-O bond formation and NO desorption. The overall downhill energetics (Figure 4) for O volatilization as NO via N attack thus serves as the fundamental mechanism behind the O abstraction by N as well as the positive correlation between N and O volatility (Figures 2 and 3). Moreover, QC findings also indicate that F and N play synergistic roles, just as earlier shown with MD. Specifically, the F attack on Si apparently assists in the N attack on O by generating additional surface *Si-O dangling bonds other than by ion bombardment, creating more active sites for N-O formation than would otherwise be available (Figure 6). This eventually leads to the observed greater volatility of both N and O when F is added to the bombardment (Figure 2). Finally, QC calculations also suggest that, vice versa, N attack on O in turn also assists in F attack on Si, facilitated by the creation of under-coordinated surface *Si following the volatilization of the NO adsorbate (Figure 4).
Figure 6.
The reaction energetics for the surface 3-coordinated *Si undergoing 2 sequential F attacks leading to the formation of 2 Si-F bonds. The most stable spin states are presented for each geometry. The potential energy sum for *Si-O (doublet) and N atom (quartet) is chosen as reference, consitently with Figures 4 and 5. The atom movements are marked with arrows for the transition state structure involving the second F attack via a subsitution pathway. The formation of another surface dangling *Si-O at the end of the 2 F attacks is high-lighted.